 2022, Vol. 54
2022, Vol. 54



2. 中国科学院大学, 北京 100049
施肥、耕作和灌溉等管理措施影响农田生态系统土壤化学性质、物理结构和生物特性,从而支持和维系农业生态功能和服务[1]。微生物占有农田生态系统中一大部分的生物量,参与几乎所有的土壤过程,直接或间接地影响土壤养分循环、土壤肥力和作物产量[2]。因此微生物群落的变化,特别是指示物种(主要指显著增加或者降低的物种)可以反映农田土壤对管理措施的动态响应和反馈[3]。过去10年中,大量工作研究了施肥对农田土壤微生物群落的影响。研究发现,施肥通过改变土壤理化性质,例如增加养分或外源碳输入改变土壤微生物群落和功能[4]。长期不施肥的土壤微生物群落以寡营养型偏好物种为主,群落功能多样但分散,而施肥后的土壤具有更多降解复杂有机质的微生物,如酸杆菌(Acidobacteria)和厚壁菌(Firmicutes)[2]。此外,不同位点的施肥指示微生物不同。位于江西南昌的长期定位试验发现,施用无机肥后显著增加放线菌(Actinobacteria)、绿弯菌(Chloroflexi)、变形菌(Proteobacteria),而有机无机配施后厚壁菌、变形菌、拟杆菌(Bacteroides)、己科河菌(Rokubacteria)和绿弯菌显著增加[5]。位于封丘的长期定位试验发现,施用无机肥会引起少数微生物丰度的剧烈波动,施用有机肥对多数微生物的影响更为温和,其中酸杆菌、拟杆菌、厚壁菌和变形菌是施有机肥后的主要响应物种,只有拟杆菌在施用无机肥后显著增加[6]。
目前对施肥响应微生物的研究多在单一田块尺度下展开,由于土壤微生物较强的地理学分布特征,导致施肥的指示物种存在争议,需要整合的、跨站点的试验调查土壤微生物对施肥响应的共性,以更全面地了解土壤微生物群落结构和功能如何随着农田管理措施而变化,并找出具有指导意义的指示物种。研究表明,虽然跨位点微生物群落结构差异巨大,但养分添加会引发全球草地生态系统微生物的一致响应,其中富营养型微生物因为竞争力强和代谢功能多样而快速生长和富集[7]。此外,施肥会降低区域尺度下微生物群落的beta多样性[8]。最新的研究发现,存在于不同位点的微生物对施肥的响应一致,尽管这样的微生物数量较少[9]。以上信息暗示了大空间尺度下施肥会引起微生物群落中特定物种(指示物种)的规律性响应。但是,也要认识到区域尺度下地理位置对微生物群落的影响大于施肥[8],即空间分异引起的微生物群落差异会干扰对施肥指示微生物的识别。因此,在阐明区域尺度下农田生态系统细菌群落对施肥的一致响应并找出相应的指示物种之前,需要降低地理分异导致的微生物群落差异。
基于此目的,本研究以中国亚热带地区的6个农业生态试验位点的水稻土为对象,在每个位点开展多年大田施肥试验,施肥处理包括CK(不施肥料)、NPK(施用含氮磷钾的矿质肥料)和OMN(施用有机肥+氮磷钾肥料)。这6个试验点覆盖了广阔的地理区域,有助于在区域尺度上了解农业生态系统中不同施肥策略下的微生物特性。研究利用Illumina高通量测序技术在得到原始微生物群落数据后,筛去带有地理特征的物种,保留不同处理下6个位点普遍存在的微生物,随后通过与不施肥处理的土壤微生物群落进行比较,以识别两种施肥策略下响应微生物群落及其变化与土壤化学性质的关系。通过比较我国亚热带区多个长期定位施肥试验土壤细菌群落,有助于识别区域尺度下2种典型施肥措施下的指示物种,以更好地利用和调控微生物来服务和指导农业生产
1 材料与方法 1.1 样品采集从中国亚热带地区的6个农业试验站采集土壤样品,6个站点的具体描述见表 1。试验点跨越约1 000 km,每个试验点都有一个施肥小区试验,设置3种处理:CK(不施肥料)、NPK(施用含氮磷钾的矿质肥料)和OMN(施用有机肥+氮磷钾肥料),各处理详细信息见表 2。样品于2014年水稻收获后采集,每个位点3种处理分别采集3个重复样本,6个位点共54个样品。样品带回实验室后,于阴凉通风处风干以去除过多水分。随后用冻存管取样分装并保存于–20℃冰箱中用于提取土壤细菌基因组DNA,剩余样品完全风干并过筛用于测定土壤基本性质。
|
|
表 1 6个野外试验位点的信息描述 |
|
|
表 2 6个试验位点不同处理的施肥情况 |
土壤pH由土水质量比1∶2.5浸提,pH计(Thermo,USA)测定[10];土壤全氮(TN)用凯氏定氮法测定;土壤全磷(TP)和全钾(TK)先用氢氟酸–高氯酸消煮,然后分别用钼蓝比色法和火焰光度法测定;土壤有效氮(AN)采用2 mol/L KCl提取,流动分析仪测定;土壤有效磷(AP)采用0.5 mol/L NaHCO3提取,钼蓝比色法测定;土壤速效钾(AK)由乙酸铵提取,由火焰光度法测定。
1.3 土壤可溶性有机质含量和组分测定称取10.00 g新鲜土壤与100 ml蒸馏水土水质量比1∶10,混合进行可溶性有机质的提取,用超声波振荡仪振荡2 h后转移到50 ml离心管中,静置后停止萃取,取出上清液过0.45 μm滤膜,滤液分为两份,一份测定浓度,另一份进行傅里叶变换离子回旋共振质谱测定可溶性有机质组分[11]。在本研究中,可溶性有机质被分为8个主要家族:脂质(0 < O/C≤0.3,1.5≤H/C≤2.5),蛋白质(0.3 < O/C≤0.55,1.5≤H/C≤2.3),氨基糖(0.55 < O/C≤0.7,1.5≤H/C≤2.2),碳水化合物(0.7 < O/C≤1.5,1.5≤H/C≤2.5),不饱和烃(0≤O/C≤0.125,0.8≤H/C≤1.5),木质素(0.125≤O/C≤0.65,0.8≤H/C≤1.5),单宁(0.65 < O/C≤1.1,0.8≤H/C≤1.5)和缩合烃(0≤O/C≤0.95,0.2≤H/C≤0.8)。
1.4 土壤DNA提取土壤样品DNA依照土壤基因组DNA提取试剂盒(MP Biochemicals, Solon, OH, USA)使用步骤提取。提取的基因组DNA溶解于50 μl TE(tris-EDTA)缓冲液中,并用分光光度计定量和凝胶电泳进行质量评价。之后,采用核酸定量仪NanoDrop ND-2000 (Thermo Scientific,Wilmington,DE,USA)对DNA进行定量检测。
1.5 16S rRNA基因PCR扩增及高通量测序本研究使用引物519F(3′-CAGCMGCCGCGGT AATWC-5′)和907R(3′-CCGTCAATTCMTTTRAGTT T-5′) 对细菌16S rRNA基因V4 ~ V5可变区进行扩增。正向引物3′端加入5 bp已知碱基序列的分类标签(Barcode) 以区分不同样品。50 μl PCR体系包括:1.25 μmol/L的dNTP,2 μl (15 μmol/L)正向和反向引物,2 μmol/L TaqDNA聚合酶(TaKaRa, Japan)和1 μl (50 ng)的DNA模板。所有样品扩增3份后混合检测以消除PCR过程中的偏好性和随机性误差,阴性对照以无菌水代替DNA模板。PCR扩增条件如下:94℃ 5 min,30个循环(94℃ 30 s,55℃ 30 s,72℃ 45 s),最后72℃延长10 min。所有PCR产物均通过1% 琼脂糖凝胶进行条带特异性检测。使用QIAquick PCR Purification Kit(Qiagen)纯化试剂盒纯化PCR扩增产物,并用NanoDrop ND-2000测定产物浓度。将不同样品的PCR产物按照等摩尔原则进行混合,使用Illumina MiSeq测序平台进行高通量测序。16S rRNA测序基因序列使用Quantitati-ve Insights Into Microbial Ecology(QIIME)平台进行分析。质量低于25、长度小于200 bp的序列被去除,并根据Barcode序列与样品进行匹配。使用Usearch算法按照97% 的相似性对序列进行聚类并进行嵌合体去除,将样品划分为不同的OTU(操作分类单元),其中每个OTU中丰度最高的序列被选作代表序列。物种注释选择SILVA 119 database(http://www.arbsilva.de/download/archive/qiime/)数据库。将所有样品的序列统一抽平到5 370条以便于后续分析。
1.6 数据分析基于Bray-Curtis距离的非度量多纬尺度分析(NMDS)用以展示群落结构差异,置换多元方差分析(permutational multivariate analysis of variance,PERMANOVA)用以检验群落结构差异显著性,PERMDISP(多变量数据统计分析)用以展示群落内离散程度。使用R软件“vegan”包进行以上分析。利用R软件中的“DEseq2”包筛选两种施肥策略与不施肥对照相比差异表达的物种,即响应物种,其阈值设定为| log2(fold change)| > 0和校正后的P≤0.05,log2(fold change) > 0和P≤0.05表示物种显著富集,log2(fold change) < 0和P≤0.05表示物种显著下降。利用冗余分析(redundancy analysis,RDA)进行群落结构与环境因子的相关性分析,揭示驱动群落结构变化的主要环境因子。方差分析(ANOVA)用于处理间土壤理化性质和有机组分差异分析,使用SPSS16.0计算。本文所有统计分析,显著和极显著水平分别为P < 0.05和P < 0.01。
2 结果分析 2.1 物种过滤研究发现,区域尺度下地理分异是引起微生物群落差异的主要原因,施肥对微生物群落组成和结构的影响不显著[8]。但对6个位点单独分析时,发现在其中4个位点(鹰潭、常熟、惠州和桂林)施肥对微生物群落有显著影响(表 3),说明地理分异导致的微生物群落差异会掩盖施肥对微生物群落的影响。因此,在数据分析前,本研究利用过滤OTU的方法减少由地理分异引起的位点间细菌群落的巨大噪音,并挑选出不同处理下6个位点共同存在的物种。通过排除不同施肥处理下6个位点≥50% 样本(9/18)中OTU丰度≤3的稀少物种,过滤得到区域尺度下各处理普遍存在的微生物,并把它们定义为“共有微生物”。降噪后得到CK处理下159个共有OTU,NPK处理下203个共有OTU,OMN处理下211个共有OTU。
|
|
表 3 PERMANOVA展示区域和局域尺度下3种处理间水稻土细菌群落结构(Bray-Curtis距离)的差异 |
如图 1A所示,过滤后各样本的微生物群落和过滤前各样本的微生物群落具有极强的相关性(r=0.97, P < 0.01),说明过滤后的物种可以代表各个样本的主要信息。PERMANOVA结果显示(图 1B和表 3),过滤后不同施肥策略下水稻土细菌群落结构存在极显著差异(P < 0.01)。因此,过滤后的微生物可以作为区域尺度下不同施肥策略中普遍存在且关键的物种。PERMDISP结果显示,过滤后OMN处理组内物种离散程度最小,其次是NPK处理,而CK处理最大(OMN(0.317 3) < NPK(0.340 4) < CK(0.374 0)),说明施肥后6个位点的微生物群落较长期不施肥更为聚集。

|
图 1 基于Bray-Curtis距离的过滤物种(263种OTU)与原始物种(5 370种OTU)的β多样性(群落不相似性)关系(A),及过滤微生物基于Bray-Curtis距离的NMDS分析(B) |
如图 2所示,过滤后2种施肥处理,特别是无机肥配施有机肥后,多位点共有OTU比长期不施肥处理多(OMN(211) > NPK(203) > CK(159))。3种处理都存在的共有OTU有122个。CK处理特有17个OTU,NPK处理特有21个OTU,以及OMN处理特有37个OTU,说明施肥,特别是有机无机配施后区域尺度下特异性共有物种增加。

|
图 2 过滤后各处理OTU个数韦恩图 |
为了探明长期施肥对微生物群落组成和结构的具体变化,对过滤后的微生物相对丰度进行差异分析,结果如图 3A ~ 3C所示。与CK处理相比,NPK处理显著富集67种OTU,显著减少24种OTU;OMN处理显著富集88种OTU,显著减少37种OTU。此外,与NPK处理相比,OMN处理显著富集45种OTU,显著减少36种OTU。

|
(图中每个点代表一个 OTU,高于中轴线(y=0)的为显著富集的 OTU,低于中轴线的为显著减少的 OTU,中轴线附近的为差异不显著的 OTU。图A:与 CK 相比,NPK 处理下显著富集和减少的微生物;图B:与 CK 相比,OMN 处理下显著富集和减少的微生物;图C:与 NPK 相比,OMN 处理下显著富集和减少的微生物。) 图 3 不同施肥处理下显著富集和减少的OTU |
对这些显著富集的OTU进行门水平上的统计(图 4A),发现施肥均显著富集绿弯菌(Chloroflexi)、变形菌(Proteobacteria)、酸杆菌(Acidobacteria)、绿菌(Chlorobi)、硝化螺旋菌(Nitrospirae)、蓝菌(Cyanobacteria)、芽单胞菌(Gemmatimonadetes)、浮霉菌(Planctomycetes)、拟杆菌(Bacteroidetes)。其中,绿弯菌、变形菌、酸杆菌、绿菌、硝化螺旋菌是主要的富集类群,分别占NPK处理和OMN处理下富集微生物的79.41% 和80.66%。NPK处理下这5种细菌门相对丰度排序为绿弯菌 > 变形菌 > 酸杆菌 > 绿菌 > 硝化螺旋菌,但OMN处理的排序为变形菌 > 绿弯菌 > 酸杆菌 > 硝化螺旋菌 > 绿菌。进一步在更高的分辨率下识别不同施肥处理的响应物种,施肥尤其是OMN处理下显著富集的绿弯菌以厌氧绳菌(Anaerolineae)为主(图 4B)。施肥显著富集的变形菌以δ-变形菌(Gammaproteobacteria)和β-变形菌(Betaproteobacteria)为主,NPK处理富集的δ-变形菌最多,但OMN处理下富集的β-变形菌最多(图 4C)。施肥尤其是OMN处理下显著富集的酸杆菌门以酸杆菌纲(Acidobacteria)为主(图 4D)。施肥显著富集的绿菌门以绿菌纲(Chlorobia)和褐藻菌纲(Ignavibacteria)为主,且OMN处理的褐藻菌相对丰度更高(图 4E)。在两种施肥策略下富集的硝化螺旋菌都属于硝化螺旋菌纲(Nitrospira),且OMN处理下富集数量和比例更多(图 4F)。此外,OMN处理的拟杆菌富集程度明显高于NPK处理(图 4G),厚壁菌是其特有的富集门类(图 4H)。

|
(A:门水平下显著富集的细菌组成;B、C、D、E、F、G:纲水平下富集的绿弯菌、变形菌、酸杆菌、绿菌、硝化螺旋菌、拟杆菌、厚壁菌物种组成) 图 4 不同施肥处理主要富集的细菌门和纲水平物种组成 |
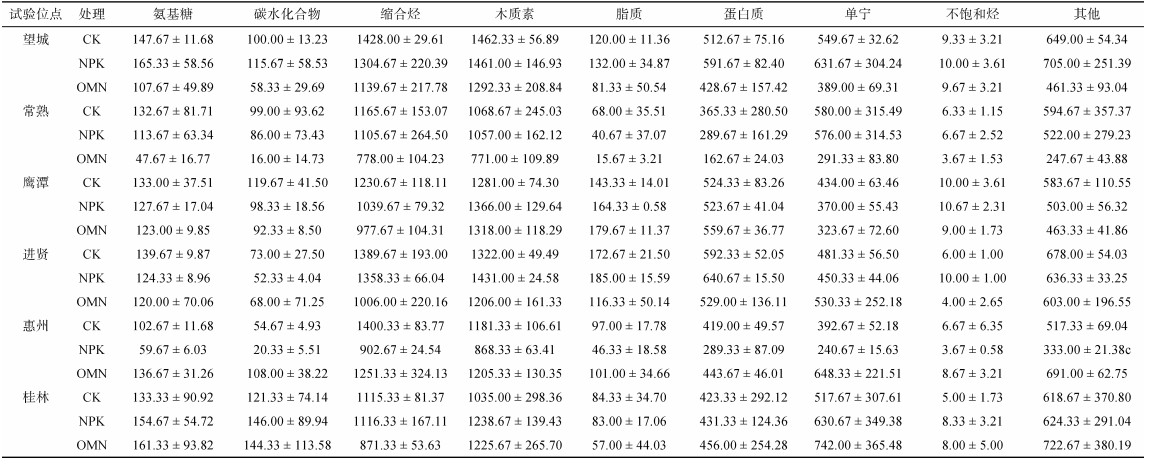
将差异分析得到的施肥响应微生物与土壤化学性质以及有机质组分进行RDA分析,以探明三者的相关性,结果如图 5A所示。不同处理下的响应微生物各自聚集在一起,并在主坐标1 (RDA1) 上明显分开,其中NPK处理和OMN处理的微生物群落分离程度较小。不同处理下的响应微生物群落与土壤性质和有机质组分呈现不一样的关系,全氮、有效磷、有机质、缩合烃含量以及pH与响应微生物群落相对丰度的相关性最强:施肥后土壤全氮、有效磷和有机质含量与两个施肥处理的响应微生物群落相对丰度呈正相关,缩合烃含量、pH与长期不施肥处理土壤中的微生物群落相对丰度呈正相关。对5种影响因子的总体(图 5B)和单个位点(表 4和表 5)进行统计,发现全氮、有效磷、有机质含量在施肥后尤其是有机无机配施后增加,而缩合烃含量、pH在施肥后有降低的趋势。尽管惠州站点的土壤有效磷在施肥后降低和进贤、鹰潭站点的土壤pH在施肥后略微升高,但不影响总体趋势。

|
(柱图上方不同小写字母表示不同处理间差异显著(P < 0.05)) 图 5 不同处理下响应细菌群落与土壤化学性质和有机组分相关性的RDA排序图(A),及不同处理下与响应微生物相关的5种环境因子经标准化后的均值(B) |
|
|
表 4 6个试验位点不同处理下土壤基本理化性质 |
|
|
表 5 6个试验位点不同处理下土壤有机质化学组分(mg/ke) |
种库假说认为,一个地区的群落组成受到区域种库的限制,区域种库由地质历史过程、物种形成、迁移扩散和消亡等决定,因此群落组成一般具有特定的地理分布格局[12]。处于不同地理位置的微生物群落因种库差异存在难以忽视的不同。本研究的过滤方法即意在减小这种不同。虽然可能造成样本微生物群落部分信息损失,如在某些位点相对丰度高且对施肥敏感的物种可能因别的位点不存在或者稀少而被排除在外,但降噪处理后的水稻土细菌群落与原始群落具有显著且强烈的相关性(图 1A),说明本研究对微生物群落的信息提取是可靠的。降噪处理后分别得到3种处理下6个位点普遍存在的细菌群落,且这些群落存在显著差异(图 1B和表 3),说明施肥会引起特定微生物的响应。揭示这些微生物对施肥的响应有助于揭示施肥策略与农田生态系统的关系。
NMDS图和PERMDISP结果均表明,与不施肥处理相比,施肥后6个位点的水稻土细菌群落更为聚集,施肥尤其是有机无机配施可降低水稻土细菌群落的空间变异。不同施肥处理过滤得到的共有微生物数量不一致,施肥尤其是有机无机配施可增加位点间共有物种(图 2),意味着施肥后尤其是有机无机配施可刺激同种微生物[8],进而增加了多样性。在3种处理中都存在的共有微生物(122个OTU)占据总过滤微生物的46.39%,这部分微生物作为核心微生物在不同处理下普遍存在,可能与维持农田生态系统的基本功能有关。
3.2 区域尺度下水稻土响应微生物与土壤化学性质和有机质组分的关系5种门类微生物在施肥后显著富集(图 4A),意味着区域尺度下施肥会引起微生物群落一致的响应。这些微生物的变化与土壤养分状况以及有机质组分的变化有关(图 5)。一方面,施肥通过改善养分条件影响微生物群落组成。施肥尤其是无机肥配施有机肥后土壤中氮、磷养分充足(图 5B和表 4),吸引偏好富营养环境的微生物生长并积极发挥生态功能,β-变形菌、硝化螺旋菌等微生物大量富集(图 4C和4F)。另一方面,施肥通过改变有机质含量和组分影响微生物群落组成(图 5B和表 5),良好的有机质分配比例促进微生物群落生长代谢和功能维持,如厌氧绳菌、褐藻菌在配施有机肥后显著富集(图 4B和4E)。缩合烃是土壤有机质中的一种热稳定化合物,性质稳定不易被微生物降解和利用[13],在长期不施肥的土壤中相对比例不断提高(表 5)。施肥后,尤其是有机无机配施后水稻土缩合烃相对含量不断降低,这可能有两个原因。其一,由于有机肥自带有机质,添加进土壤后会改变有机质结构,难降解有机质相对比例降低;其二,施肥富集的微生物可发挥多种降解功能,加强了对难降解组分的利用,有机质组分进一步得到平衡。因此,施肥,特别是施用有机肥通过影响土壤有机质和养分导致微生物群落的一致变化。
土壤pH可对微生物群落产生强烈影响,常被视为细菌群落组成的良好预测因子[14]。本研究中pH虽然不是首要预测因子,但施肥后土壤pH下降(图 5B和表 4),可能是酸杆菌在施肥后显著富集的原因[15]。拟杆菌、变形菌等微生物与pH相关性并无准确定论[16],说明单一因子并不能完全解释土壤细菌群落的变化。综上,不同微生物对不同土壤性质的敏感度不同,是土壤性质的特定变化以及变化幅度而不一定是施肥本身可能更好地预测特定景观中微生物群落组成的变化[15],这是不同施肥后微生物群落变化的原因,可能也是不同环境条件下同种施肥策略研究结论不一致的原因,同时也说明了大尺度研究的必要性。
3.3 区域尺度下响应施肥的水稻土指示物种区域尺度下,微生物对施肥的响应存在一致规律。在门水平上,两种施肥策略下富集的微生物主要是绿弯菌、变形菌、酸杆菌、绿菌和硝化螺旋菌(图 4A),可作为水稻土对施肥响应的指示微生物。但在更高分辨率的分类水平上,两种施肥策略的响应微生物存在差异(图 4B ~ 4H)。有机无机配施处理下,β-变形菌、酸杆菌、硝化螺旋菌富集程度更高(图 4C),这与施肥导致土壤化学性质变化有关。变形菌是水稻土的优势菌群之一[17-18],β-变形菌对富营养环境十分敏感,养分充足时会大量生长[19]。研究发现酸杆菌相对丰度和土壤pH呈显著负相关关系[15],这与本研究施肥后水稻土pH呈下降趋势的结论一致(图 5B和表 4)。此外,也有研究发现,一些酸杆菌亚群(Acidobacteria Gp4和Gp6)在有机碳含量高的土壤中非常丰富[20],这可能是有机无机配施富集酸杆菌程度更高的原因。硝化螺旋菌门在土壤亚硝酸盐氧化过程中起重要作用[21],有机无机配施的土壤氮有效性增加幅度更高(图 5B和表 4),有助于硝化螺旋菌大量繁殖,进而提高氮素的转化效率,促进氮元素在土壤与作物间的循环和流通。褐藻菌和厌氧绳菌在有机无机配施下增加更为明显(图 4B和4E),它们的响应可能与土壤有机质转化密切相关。本研究有机肥中秸秆的主要成分为纤维素、半纤维素(表 2),可诱导褐藻菌和厌氧绳菌分泌纤维素酶、半纤维素酶、碳水化合物酯酶和多糖裂解酶[22-23],促进纤维素降解等有机质转化过程。拟杆菌和厚壁菌是有机无机配施后显著富集的特定微生物(图 4G和4H),除了能在富营养环境中快速生长[24],它们还参与多种代谢途径促进有机质降解[25]。此外,有研究发现,长期施用田间污泥后,土壤中拟杆菌和厚壁菌与抗性基因增加具有显著相关性[26]。通过分泌抗生素抑制病原菌的生长繁殖,拟杆菌和厚壁菌有助于健康土壤系统的形成和维持[27],可以作为健康土壤的指示微生物。这两类微生物的增加有利于农田生态系统的可持续发展,说明有机无机配施是环境友好型的田间管理措施。
4 结论在过滤排除土壤微生物自身地理学分布引起的变异度后,本研究系统评估了区域尺度下不同施肥对水稻土细菌群落的影响。研究发现,区域尺度下水稻土细菌群落对施肥策略具有一致的响应规律。长期施肥可增加土壤全氮、有效磷和有机质含量,使得不同位点的土壤微生物群落发生趋于一致的变化,指示微生物绿弯菌、变形菌、酸杆菌、硝化螺旋菌和绿菌的相对丰度显著增加。有机无机配施进一步增加了与有机质转化相关以及富营养型的β-变形菌、厌氧绳菌、褐藻菌、拟杆菌和厚壁菌。这些指示微生物在土壤性质改善和土壤生态保持方面发挥积极的功能,它们的丰度和多样性影响碳循环、氮循环过程和农业生产,尤其有机无机配施处理下,特定的指示微生物拟杆菌、厚壁菌对维持土壤健康有重要的积极意义。
| [1] |
张婷婷, 王丽芳, 张德健, 等. 土壤管理措施及环境因素对农田土壤微生物的影响[J]. 北方农业学报, 2020, 48(6): 87-92 (  0) 0) |
| [2] |
Hartmann M, Frey B, Mayer J, et al. Distinct soil microbial diversity under long-term organic and conventional farming[J]. The ISME Journal, 2015, 9(5): 1177-1194 DOI:10.1038/ismej.2014.210 ( 0) |
| [3] |
Barrios E. Soil biota, ecosystem services and land productivity[J]. Ecological Economics, 2007, 64(2): 269-285 DOI:10.1016/j.ecolecon.2007.03.004 ( 0) |
| [4] |
Zhong W H, Gu T, Wang W, et al. The effects of mineral fertilizer and organic manure on soil microbial community and diversity[J]. Plant and Soil, 2010, 326(1/2): 511-522 ( 0) |
| [5] |
Liu J A, Shu A P, Song W F, et al. Long-term organic fertilizer substitution increases rice yield by improving soil properties and regulating soil bacteria[J]. Geoderma, 2021, 404: 115287 DOI:10.1016/j.geoderma.2021.115287 ( 0) |
| [6] |
Yao T Y, Chen R R, Zhang J W, et al. Divergent patterns of microbial community composition shift under two fertilization regimes revealed by responding species[J]. Applied Soil Ecology, 2020, 154: 103590 DOI:10.1016/j.apsoil.2020.103590 ( 0) |
| [7] |
Leff J W, Jones S E, Prober S M, et al. Consistent responses of soil microbial communities to elevated nutrient inputs in grasslands across the globe[J]. Proceedings of the National Academy of Sciences of the United States of America, 2015, 112(35): 10967-10972 DOI:10.1073/pnas.1508382112 ( 0) |
| [8] |
Chen R R, Zhong L H, Jing Z W, et al. Fertilization decreases compositional variation of paddy bacterial community across geographical gradient[J]. Soil Biology and Biochemistry, 2017, 114: 181-188 DOI:10.1016/j.soilbio.2017.07.013 ( 0) |
| [9] |
Feng Y Z, Delgado-Baquerizo M, Zhu Y G, et al. Responses of soil bacterial diversity to fertilization are driven by local environmental context across China[J]. Engineering, 2022, 12: 164-170 DOI:10.1016/j.eng.2021.09.012 ( 0) |
| [10] |
中国科学院南京土壤研究所. 土壤理化分析[M].
上海科学技术出版社, 上海, 1978
( 0) |
| [11] |
Zhang J W, Feng Y Z, Wu M, et al. Evaluation of microbe-driven soil organic matter quantity and quality by thermodynamic theory[J]. mBio, 2021, 12(1): e03252-e03220 ( 0) |
| [12] |
Zobel M. The relative of species pools in determining plant species richness: An alternative explanation of species coexistence?[J]. Trends in Ecology & Evolution, 1997, 12(7): 266-269 ( 0) |
| [13] |
Solihat N N, Yustiawati, Kim S, et al. Elucidating molecular level impact of peat fire on soil organic matter by laser desorption ionization Fourier transform ion cyclotron resonance mass spectrometry[J]. Analytical and Bioanalytical Chemistry, 2019, 411(27): 7303-7313 DOI:10.1007/s00216-019-02108-2 ( 0) |
| [14] |
Rousk J, Bååth E, Brookes P C, et al. Soil bacterial and fungal communities across a pH gradient in an arable soil[J]. The ISME Journal, 2010, 4(10): 1340-1351 DOI:10.1038/ismej.2010.58 ( 0) |
| [15] |
Lauber C L, Strickland M S, Bradford M A, et al. The influence of soil properties on the structure of bacterial and fungal communities across land-use types[J]. Soil Biology and Biochemistry, 2008, 40(9): 2407-2415 DOI:10.1016/j.soilbio.2008.05.021 ( 0) |
| [16] |
Zeng J, Liu X J, Song L, et al. Nitrogen fertilization directly affects soil bacterial diversity and indirectly affects bacterial community composition[J]. Soil Biology and Biochemistry, 2016, 92: 41-49 DOI:10.1016/j.soilbio.2015.09.018 ( 0) |
| [17] |
Zhu J, Peng H, Ji X H, et al. Effects of reduced inorganic fertilization and rice straw recovery on soil enzyme activities and bacterial community in double-rice paddy soils[J]. European Journal of Soil Biology, 2019, 94: 103116 DOI:10.1016/j.ejsobi.2019.103116 ( 0) |
| [18] |
Liu L, Ding M J, Zhou L K, et al. Effects of different rice straw on soil microbial community structure[J]. Agronomy Journal, 2021, 113(2): 794-805 DOI:10.1002/agj2.20509 ( 0) |
| [19] |
Wang J J, Ma Y, Di L, et al. Straw incorporation with nitrogen amendment shapes bacterial community structure in an iron-rich paddy soil by altering nitrogen reserves[J]. Microorganisms, 2021, 9(5): 988 DOI:10.3390/microorganisms9050988 ( 0) |
| [20] |
Sul W J, Asuming-Brempong S, Wang Q, et al. Tropical agricultural land management influences on soil microbial communities through its effect on soil organic carbon[J]. Soil Biology and Biochemistry, 2013, 65: 33-38 ( 0) |
| [21] |
Li Y Y, Chen L Q, Wen H Y, et al. 454 pyrosequencing analysis of bacterial diversity revealed by a comparative study of soils from mining subsidence and reclamation areas[J]. Journal of Microbiology and Biotechnology, 2014, 24(3): 313-323 ( 0) |
| [22] |
Podosokorskaya O A, Bonch-Osmolovskaya E A, Novikov A A, et al. Ornatilinea apprima gen. nov., sp. nov., a cellulolytic representative of the class Anaerolineae[J]. International Journal of Systematic and Evolutionary Microbiology, 2013, 63: 86-92 ( 0) |
| [23] |
Bei Q C, Peng J J, Liesack W. Shedding light on the functional role of the Ignavibacteria in Italian rice field soil: A meta-genomic/transcriptomic analysis[J]. Soil Biology and Biochemistry, 2021, 163: 108444 ( 0) |
| [24] |
Shahi A, Aydin S, Ince B, et al. Reconstruction of bacterial community structure and variation for enhanced petroleum hydrocarbons degradation through biostimulation of oil contaminated soil[J]. Chemical Engineering Journal, 2016, 306: 60-66 ( 0) |
| [25] |
Do T H, Dao T K, Nguyen K H V, et al. Metagenomic analysis of bacterial community structure and diversity of lignocellulolytic bacteria in Vietnamese native goat rumen[J]. Asian-Australasian Journal of Animal Sciences, 2018, 31(5): 738-747 ( 0) |
| [26] |
Bulgarelli D, Schlaeppi K, Spaepen S, et al. Structure and functions of the bacterial microbiota of plants[J]. Annual Review of Plant Biology, 2013, 64: 807-838 ( 0) |
| [27] |
Salamat S S, Hassan M A, Shirai Y, et al. Effect of inorganic fertilizer application on soil microbial diversity in an oil palm plantation[J]. BioResources, 2021, 16(2): 2279-2302 ( 0) |
2. University of Chinese Academy of Sciences, Beijing 100049, China